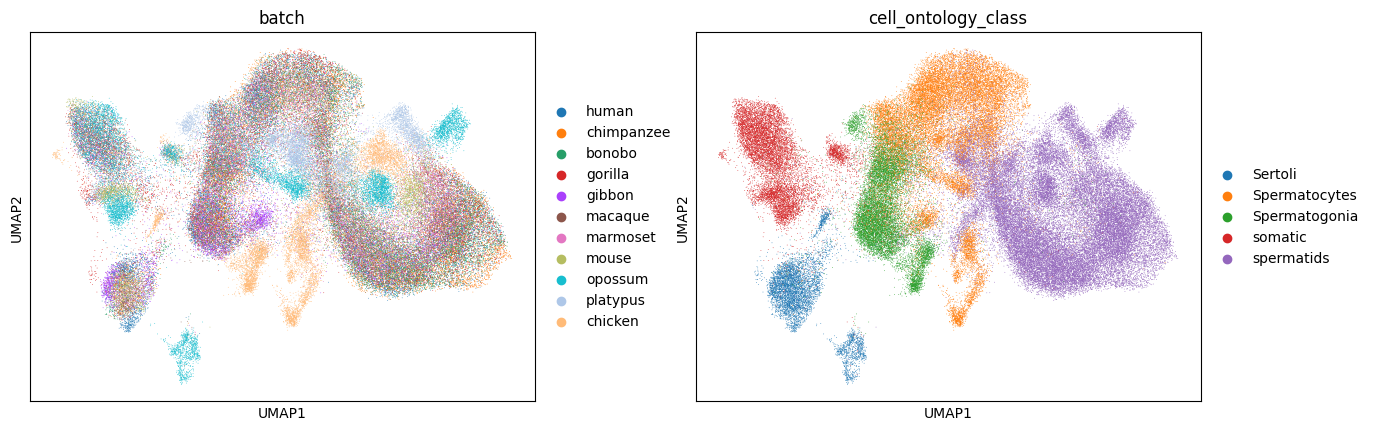
CAMEX integrates testis scRNA-seq dataset across 11 species
This tutorial demonstrates that CAMEX uncovers the conserved differentiation process in the testis across 11 species.
Here, we use a testis scRNA-seq dataset across 11 species. Processed h5ad files can be downloaded from https://drive.google.com/drive/folders/1rwdjEvWFEFw82a0x2JzMi2jXICbUc5eb?usp=sharing
[1]:
import warnings
warnings.filterwarnings("ignore")
[2]:
import os
import time
import torch
import shutil
import warnings
import argparse
import importlib
import scanpy as sc
import pandas as pd
import numpy as mp
from CAMEX.base import Dataset
from CAMEX.trainer import Trainer
[2]:
from params import PARAMS
[3]:
t1 = time.time()
make log dir
[4]:
time_start = time.strftime("%Y-%m-%d-%H-%M-%S")
log_path = f'./log/{time_start}/'
for k, v in PARAMS.items():
v['time_start'] = time_start
v['log_path'] = log_path
print(log_path)
./log/2024-04-12-17-02-43/
[5]:
os.makedirs(log_path, exist_ok=True)
shutil.copy('params.py', log_path + 'params_current.py')
print(f'time: {time_start}')
time: 2024-04-12-17-02-43
preprocess scRNA_seq data to construct a heterogeneous graph of cells and genes
[6]:
# —————————————————————————————————— 1 preprocess
print('start preprocess')
dataset = Dataset(**PARAMS['preprocess'])
adata_CAMEX = dataset.adata_whole
dgl_data = dataset.dgl_data
start preprocess
raw-testis-human-Murat: reference raw-testis-chimpanzee-Murat: query raw-testis-bonobo-Murat: query raw-testis-gorilla-Murat: query raw-testis-gibbon-Murat: query raw-testis-macaque-Murat: query raw-testis-marmoset-Murat: query raw-testis-mouse-Murat: query raw-testis-opossum-Murat: query raw-testis-platypus-Murat: query raw-testis-chicken-Murat: query
spermatids 6359 6044 5452 3144 2050 2794 3266 2629 3475 4500.0 3501
Spermatocytes 3878 3210 2802 1640 1932 1781 1855 433 1564 1443.0 2566
Spermatogonia 3025 2021 1217 1237 978 784 872 257 569 542.0 1935
somatic 1394 432 150 1098 617 1686 216 1438 3394 266.0 148
Sertoli 1355 622 252 204 995 573 175 507 924 NaN 263
[7]:
print('start train')
trainer = Trainer(adata_CAMEX, dgl_data, **PARAMS['train'])
start train
integration
[8]:
trainer.integration()
--------------------------------------------- integration ---------------------------------------------
epoch: 0, loss: 165.27696962284864
epoch: 1, loss: 76.95408240296787
epoch: 2, loss: 76.09822329184166
epoch: 3, loss: 75.63389971740264
epoch: 4, loss: 75.28416913254817
epoch: 5, loss: 75.07050793870052
epoch: 6, loss: 74.85558778002746
epoch: 7, loss: 74.74335967329212
epoch: 8, loss: 74.53497951192067
epoch: 9, loss: 74.40225363136234
[9]:
adata_CAMEX.write_h5ad(log_path + 'adata_CAMEX.h5ad', compression='gzip')
[10]:
t2 = time.time()
[11]:
print(f'time usage: {round(t2-t1)} seconds')
time usage: 1819 seconds
analysis
[12]:
log_path
[12]:
'./log/2024-04-12-17-02-43/'
[13]:
adata_CAMEX = sc.read_h5ad(log_path + 'adata_CAMEX.h5ad')
adata_CAMEX
[13]:
AnnData object with n_obs × n_vars = 96464 × 2000
obs: 'sample_batch', 'cell_type', 'cell_ontology_class', 'batch', 'n_genes_by_counts', 'total_counts', 'cell_ontology_class_num', 'cell_class', 'cell_class_num'
var: 'n_cells_by_counts', 'mean_counts', 'pct_dropout_by_counts', 'total_counts', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection', 'mean', 'std'
uns: 'cell_type', 'data_order', 'dataset_description', 'dataset_type', 'hvg', 'log1p', 'neighbors', 'pca'
obsm: 'X_CAMEX_Integration', 'X_pca'
varm: 'PCs'
layers: 'counts'
obsp: 'connectivities', 'distances'
[14]:
adata_CAMEX.obsm['X_CAMEX_Integration'].shape
[14]:
(96464, 128)
[15]:
sc.pp.neighbors(adata_CAMEX, use_rep='X_CAMEX_Integration')
[16]:
sc.tl.umap(adata_CAMEX)
[17]:
sc.tl.leiden(adata_CAMEX, resolution=0.2)
[18]:
sc.pl.umap(adata_CAMEX, color=['batch', 'cell_ontology_class', 'leiden'], wspace=0.6)

Manually concatenate the 11 species scRNA-seq datasets and utilize Harmony for integration.
human
[2]:
adata_human = sc.read_h5ad('./dataset/raw-testis-human-Murat.h5ad')
adata_human
[2]:
AnnData object with n_obs × n_vars = 16011 × 26186
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[3]:
adata_human.var_names_make_unique()
adata_human.obs_names_make_unique()
[4]:
adata_human.X.max()
[4]:
1409.0
chimpanzee
[5]:
adata_chimpanzee = sc.read_h5ad('./dataset/raw-testis-chimpanzee-Murat.h5ad')
adata_chimpanzee
[5]:
AnnData object with n_obs × n_vars = 12329 × 14705
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[6]:
gene_array = pd.read_csv('./dataset/gene_matches_human2chimpanzee.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_chimpanzee.var.index) & set(gene_dict.keys()))
[7]:
adata_chimpanzee = adata_chimpanzee[:, gene_sub].copy()
adata_chimpanzee
[7]:
AnnData object with n_obs × n_vars = 12329 × 14455
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[8]:
adata_chimpanzee.var.index = adata_chimpanzee.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_chimpanzee
[8]:
AnnData object with n_obs × n_vars = 12329 × 14455
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[9]:
adata_chimpanzee.var_names_make_unique()
adata_chimpanzee.obs_names_make_unique()
bonobo
[10]:
adata_bonobo = sc.read_h5ad('./dataset/raw-testis-bonobo-Murat.h5ad')
adata_bonobo
[10]:
AnnData object with n_obs × n_vars = 9873 × 14743
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[11]:
gene_array = pd.read_csv('./dataset/gene_matches_human2bonobo.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_bonobo.var.index) & set(gene_dict.keys()))
[12]:
adata_bonobo = adata_bonobo[:, gene_sub].copy()
adata_bonobo
[12]:
AnnData object with n_obs × n_vars = 9873 × 12843
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[13]:
adata_bonobo.var.index = adata_bonobo.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_bonobo
[13]:
AnnData object with n_obs × n_vars = 9873 × 12843
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[14]:
adata_bonobo.var_names_make_unique()
adata_bonobo.obs_names_make_unique()
gorilla
[15]:
adata_gorilla = sc.read_h5ad('./dataset/raw-testis-gorilla-Murat.h5ad')
adata_gorilla
[15]:
AnnData object with n_obs × n_vars = 7323 × 14717
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[16]:
gene_array = pd.read_csv('./dataset/gene_matches_human2gorilla.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_gorilla.var.index) & set(gene_dict.keys()))
[17]:
adata_gorilla = adata_gorilla[:, gene_sub].copy()
adata_gorilla
[17]:
AnnData object with n_obs × n_vars = 7323 × 14680
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[18]:
adata_gorilla.var.index = adata_gorilla.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_gorilla
[18]:
AnnData object with n_obs × n_vars = 7323 × 14680
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[19]:
adata_gorilla.var_names_make_unique()
adata_gorilla.obs_names_make_unique()
gibbon
[20]:
adata_gibbon = sc.read_h5ad('./dataset/raw-testis-gibbon-Murat.h5ad')
adata_gibbon
[20]:
AnnData object with n_obs × n_vars = 6572 × 13419
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[21]:
gene_array = pd.read_csv('./dataset/gene_matches_human2gibbon.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_gibbon.var.index) & set(gene_dict.keys()))
[22]:
adata_gibbon = adata_gibbon[:, gene_sub].copy()
adata_gibbon
[22]:
AnnData object with n_obs × n_vars = 6572 × 13416
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[23]:
adata_gibbon.var.index = adata_gibbon.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_gibbon
[23]:
AnnData object with n_obs × n_vars = 6572 × 13416
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[24]:
adata_gibbon.var_names_make_unique()
adata_gibbon.obs_names_make_unique()
macaque
[25]:
adata_macaque = sc.read_h5ad('./dataset/raw-testis-macaque-Murat.h5ad')
adata_macaque
[25]:
AnnData object with n_obs × n_vars = 7618 × 13333
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[26]:
gene_array = pd.read_csv('./dataset/gene_matches_human2macaque.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_macaque.var.index) & set(gene_dict.keys()))
[27]:
adata_macaque = adata_macaque[:, gene_sub].copy()
adata_macaque
[27]:
AnnData object with n_obs × n_vars = 7618 × 12842
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[28]:
adata_macaque.var.index = adata_macaque.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_macaque
[28]:
AnnData object with n_obs × n_vars = 7618 × 12842
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[29]:
adata_macaque.var_names_make_unique()
adata_macaque.obs_names_make_unique()
marmoset
[30]:
adata_marmoset = sc.read_h5ad('./dataset/raw-testis-marmoset-Murat.h5ad')
adata_marmoset
[30]:
AnnData object with n_obs × n_vars = 6384 × 11078
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[31]:
gene_array = pd.read_csv('./dataset/gene_matches_human2marmoset.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_marmoset.var.index) & set(gene_dict.keys()))
[32]:
adata_marmoset = adata_marmoset[:, gene_sub].copy()
adata_marmoset
[32]:
AnnData object with n_obs × n_vars = 6384 × 11041
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[33]:
adata_marmoset.var.index = adata_marmoset.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_marmoset
[33]:
AnnData object with n_obs × n_vars = 6384 × 11041
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[34]:
adata_marmoset.var_names_make_unique()
adata_marmoset.obs_names_make_unique()
mouse
[35]:
adata_mouse = sc.read_h5ad('./dataset/raw-testis-mouse-Murat.h5ad')
adata_mouse
[35]:
AnnData object with n_obs × n_vars = 5264 × 24208
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[36]:
gene_array = pd.read_csv('./dataset/gene_matches_human2mouse.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_mouse.var.index) & set(gene_dict.keys()))
[37]:
adata_mouse = adata_mouse[:, gene_sub].copy()
adata_mouse
[37]:
AnnData object with n_obs × n_vars = 5264 × 15618
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[38]:
adata_mouse.var.index = adata_mouse.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_mouse
[38]:
AnnData object with n_obs × n_vars = 5264 × 15618
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[39]:
adata_mouse.var_names_make_unique()
adata_mouse.obs_names_make_unique()
opossum
[40]:
adata_opossum = sc.read_h5ad('./dataset/raw-testis-opossum-Murat.h5ad')
adata_opossum
[40]:
AnnData object with n_obs × n_vars = 9926 × 9697
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[41]:
gene_array = pd.read_csv('./dataset/gene_matches_human2opossum.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_opossum.var.index) & set(gene_dict.keys()))
[42]:
adata_opossum = adata_opossum[:, gene_sub].copy()
adata_opossum
[42]:
AnnData object with n_obs × n_vars = 9926 × 9670
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[43]:
adata_opossum.var.index = adata_opossum.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_opossum
[43]:
AnnData object with n_obs × n_vars = 9926 × 9670
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[44]:
adata_opossum.var_names_make_unique()
adata_opossum.obs_names_make_unique()
platypus
[45]:
adata_platypus = sc.read_h5ad('./dataset/raw-testis-platypus-Murat.h5ad')
adata_platypus
[45]:
AnnData object with n_obs × n_vars = 6751 × 11790
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[46]:
gene_array = pd.read_csv('./dataset/gene_matches_human2platypus.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_platypus.var.index) & set(gene_dict.keys()))
[47]:
adata_platypus = adata_platypus[:, gene_sub].copy()
adata_platypus
[47]:
AnnData object with n_obs × n_vars = 6751 × 11434
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[48]:
adata_platypus.var.index = adata_platypus.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_platypus
[48]:
AnnData object with n_obs × n_vars = 6751 × 11434
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[49]:
adata_platypus.var_names_make_unique()
adata_platypus.obs_names_make_unique()
chicken
[50]:
adata_chicken = sc.read_h5ad('./dataset/raw-testis-chicken-Murat.h5ad')
adata_chicken
[50]:
AnnData object with n_obs × n_vars = 8413 × 10725
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[51]:
gene_array = pd.read_csv('./dataset/gene_matches_human2chicken.csv').to_numpy()
gene_dict = {item[1]: item[0] for item in gene_array}
gene_sub = list(set(adata_chicken.var.index) & set(gene_dict.keys()))
[52]:
adata_chicken = adata_chicken[:, gene_sub].copy()
adata_chicken
[52]:
AnnData object with n_obs × n_vars = 8413 × 9333
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[53]:
adata_chicken.var.index = adata_chicken.var.loc[:, 'gene_symbol'].apply(lambda x: gene_dict[x])
adata_chicken
[53]:
AnnData object with n_obs × n_vars = 8413 × 9333
obs: 'sample_batch', 'cell_type', 'cell_ontology_class'
var: 'gene', 'gene_symbol'
[54]:
adata_chicken.var_names_make_unique()
adata_chicken.obs_names_make_unique()
concat
[58]:
adata_11species = sc.concat([adata_human, adata_chimpanzee, adata_bonobo, adata_gorilla, adata_gibbon, adata_macaque, adata_marmoset,
adata_mouse, adata_opossum, adata_platypus, adata_chicken], label='batch',
keys=['human', 'chimpanzee', 'bonobo', 'gorilla', 'gibbon', 'macaque', 'marmoset', 'mouse','opossum', 'platypus', 'chicken'])
[60]:
adata_11species
[60]:
AnnData object with n_obs × n_vars = 96464 × 3048
obs: 'sample_batch', 'cell_type', 'cell_ontology_class', 'batch'
[62]:
adata_11species.var.loc[:, 'symbol'] = adata_11species.var.index
[63]:
adata_11species.layers['counts'] = adata_11species.X.toarray()
adata_11species.raw = adata_11species
adata_11species.X.max(), adata_11species.layers['counts'].max()
[63]:
(351.0, 351.0)
[64]:
sc.pp.normalize_total(adata_11species)
sc.pp.log1p(adata_11species)
[66]:
sc.pp.highly_variable_genes(adata_11species, batch_key='batch', subset=True, n_top_genes=2000)
adata_11species
[66]:
AnnData object with n_obs × n_vars = 96464 × 2000
obs: 'sample_batch', 'cell_type', 'cell_ontology_class', 'batch'
var: 'symbol', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'log1p', 'hvg'
layers: 'counts'
[67]:
sc.pp.pca(adata_11species)
[68]:
sc.pp.neighbors(adata_11species)
[69]:
sc.tl.umap(adata_11species)
[70]:
sc.pl.umap(adata_11species, color=['batch', 'cell_ontology_class'], wspace=0.2)
C:\Users\10360\anaconda3\envs\py39\lib\site-packages\scanpy\plotting\_tools\scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
C:\Users\10360\anaconda3\envs\py39\lib\site-packages\scanpy\plotting\_tools\scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
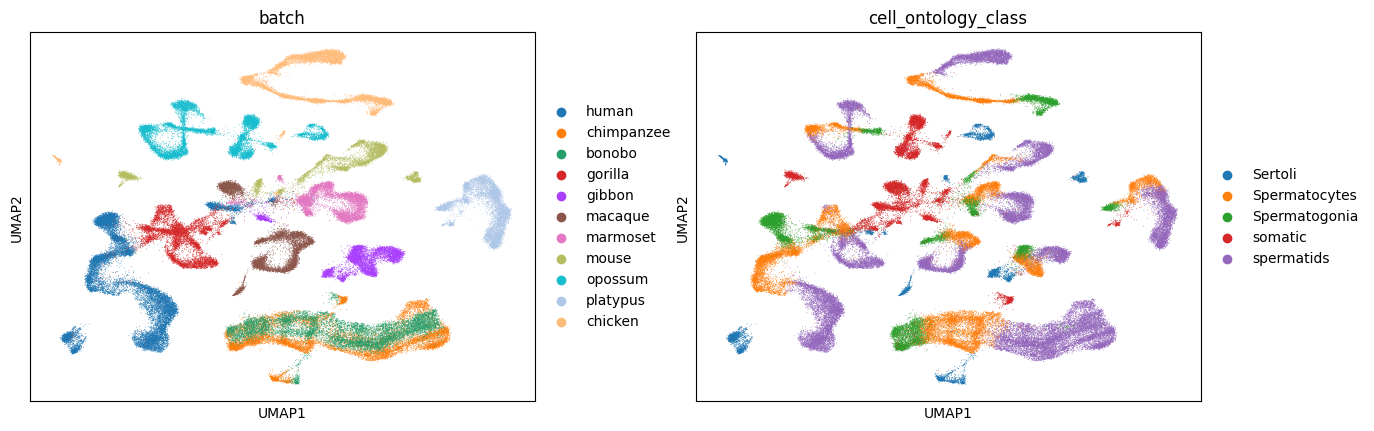
harmnoy integrate
[71]:
import scanpy.external as sce
[72]:
adata_harmony = adata_11species.copy()
adata_harmony
[72]:
AnnData object with n_obs × n_vars = 96464 × 2000
obs: 'sample_batch', 'cell_type', 'cell_ontology_class', 'batch'
var: 'symbol', 'highly_variable', 'means', 'dispersions', 'dispersions_norm', 'highly_variable_nbatches', 'highly_variable_intersection'
uns: 'log1p', 'hvg', 'pca', 'neighbors', 'umap', 'batch_colors', 'cell_ontology_class_colors'
obsm: 'X_pca', 'X_umap'
varm: 'PCs'
layers: 'counts'
obsp: 'distances', 'connectivities'
[73]:
sce.pp.harmony_integrate(adata_harmony, 'batch')
2024-03-21 08:54:49,716 - harmonypy - INFO - Computing initial centroids with sklearn.KMeans...
2024-03-21 08:55:18,106 - harmonypy - INFO - sklearn.KMeans initialization complete.
2024-03-21 08:55:18,904 - harmonypy - INFO - Iteration 1 of 10
2024-03-21 08:56:49,743 - harmonypy - INFO - Iteration 2 of 10
2024-03-21 08:58:06,853 - harmonypy - INFO - Converged after 2 iterations
[74]:
sc.pp.neighbors(adata_harmony, use_rep='X_pca_harmony')
sc.tl.umap(adata_harmony)
sc.pl.umap(adata_harmony, color=['batch', 'cell_ontology_class'], wspace=0.2)
C:\Users\10360\anaconda3\envs\py39\lib\site-packages\scanpy\plotting\_tools\scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(
C:\Users\10360\anaconda3\envs\py39\lib\site-packages\scanpy\plotting\_tools\scatterplots.py:392: UserWarning: No data for colormapping provided via 'c'. Parameters 'cmap' will be ignored
cax = scatter(